Sterimol¶
The Sterimol parameters L, B1 and B5 as described by Verloop [1][2] are implemented. Note that Sterimol parameters should be calculated with hydrogen as the dummy atom, to be consistent with values in the literature (see Background).
Module¶
The Sterimol class calculates and stores Sterimol parameters.
>>> from morfeus import Sterimol, read_xyz
>>> elements, coordinates = read_xyz("tBu.xyz")
>>> sterimol = Sterimol(elements, coordinates, 1, 2)
>>> sterimol.L_value
4.209831193078874
>>> sterimol.B_1_value
2.8650676183152837
>>> sterimol.B_5_value
3.26903261263369
>>> sterimol.print_report()
L B_1 B_5
4.21 2.87 3.27
The first given atom index argument corresponds to the index of the dummy H atom (1-indexed), while the second one corresponds to the atom bound to the dummy H.
Radii can be changed with the argument radii_type=<str> and custom radii
can be supplied as a list with radii=<list>.
The bond length between atoms 1 and 2 and the uncorrected L values (without the historical addition of 0.40 Å) can also be obtained.
>>> sterimol.L_value_uncorrected
3.8098311930788737
>>> sterimol.bond_length
1.1
>>> sterimol.print_report(verbose=True)
L B_1 B_5 L_uncorr d(a1-a2)
4.21 2.87 3.27 3.81 1.10
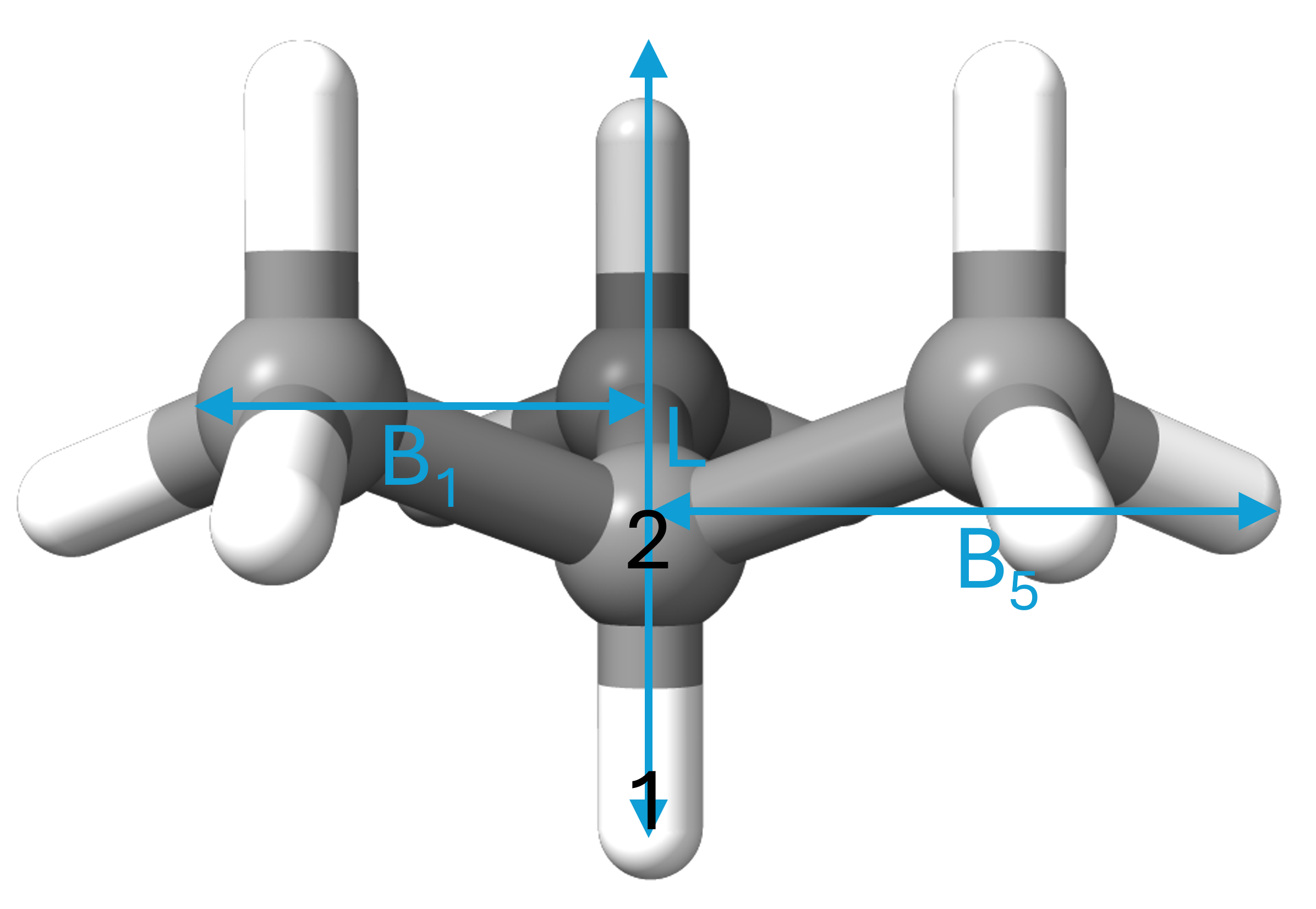
Below is illustrated the Sterimol parameters of the tert-Butyl ligand as calculated with the code above.
More information can be found with help(Sterimol) or in the API:
Sterimol.
Buried Sterimol¶
The Sterimol vectors can be “buried”:
>>> elements, coordinates = read_xyz("P_p-Tol_3.xyz")
>>> sterimol = Sterimol(elements, coordinates, 1, 2)
>>> sterimol.print_report()
L B_1 B_5
7.44 4.94 7.44
>>> sterimol.bury(method="delete")
>>> sterimol.print_report()
L B_1 B_5
6.92 4.27 6.04
>>> elements, coordinates = read_xyz("P_p-Tol_3.xyz")
>>> sterimol = Sterimol(elements, coordinates, 1, 2)
>>> sterimol.print_report()
L B_1 B_5
7.44 4.94 7.44
>>> sterimol.bury(method="truncate")
>>> sterimol.print_report()
L B_1 B_5
5.90 4.27 5.01
>>> elements, coordinates = read_xyz("P_p-Tol_3.xyz")
>>> sterimol = Sterimol(elements, coordinates, 1, 2)
>>> sterimol.print_report()
L B_1 B_5
7.44 4.94 7.44
>>> sterimol.bury(method="slice")
>>> sterimol.print_report()
L B_1 B_5
5.82 3.77 5.24
There are three different methods for doing this:
deleteAtoms outside the sphere + 0.5 vdW radius are deleted and the Sterimol vectors are calculated. This is the default.
truncateSterimol vectors are calculated as usual, but truncated in length by the sphere.
sliceA point vdW surface is constructed from the atoms and all points outside the sphere are removed. Then the Sterimol vectors are computed based on the remaining points.
A standard sphere radius of 5.5 Å is used that can be changed with
sphere_radius=<float>. For the delete method, the scaling factor for
the atom cutoff can be changed with radii_scale=<float>. For more
information, see the API:
Sterimol.bury
Command line script¶
The command line script gives access to the basic functionality from the terminal.
$ morfeus sterimol tBu.xyz - 1 2 - print_report
L B_1 B_5
4.21 2.86 3.27
Background¶
The Sterimol parameters were developed by Verloop to describe the steric size of substituents. The atom attached to the substituent in the calculation (by definition H) is called atom 1 and the first atom in the substituent is called atom 2. L can be described as the depth of the substituent. It is defined as the length of the vector going from atom 1, through atom 2 and ending on the tangent of the vdW surface. For historical reasons, L is corrected by adding 0.40 Å to this length. This was due to a shift from using C(sp2) to H as dummy atom.
B1 and B5 can be described as the minimum and maximum rotational size of the substituent. They are defined as the shortest and longest vectors from atom 2 to a tangent plane of the vdW surface which are perpendicular to the L vector, respectively.
ᴍᴏʀғᴇᴜs has been benchmarked against Paton’s Sterimol package. Using exactly the same radii (Paton’s modified Bondi), almost identical results are obtained. (Note that ᴍᴏʀғᴇᴜs normally uses 1.20 Å as the Bondi vdW radius for H). ᴍᴏʀғᴇᴜs calculates the B1 and B5 parameters by a different approach from the original code. First, atomic spheres are created with a certain density of points. B1 and B5 are then obtained by projection of atoms onto vectors spanning the whole 360 degrees in the plane perpendicular to L. B5 is obtained from the largest projection, while B1 is obtained from the smallest maximum projection for the set of vectors.
Buried Sterimol was developed by Tobias Gensch while working in the group of
Matthew Sigman at the University of Utah [3]. It is
intended to limit the Sterimol vectors to a volume of interest in the
philosophy of the buried volume. The original implementation uses the
delete algorithm. ᴍᴏʀғᴇᴜs uses the CRC handbook radii by default instead of
the modified Bondi radii in the original article, so results with the defualt
settings might be slighly different.
References