Cone angle¶
Exact cone angles are implemented as described by Allen and co-workers [1].
Module¶
The ConeAngle class is provided to calculate and store the cone angles.
>>> from morfeus import ConeAngle, read_xyz
>>> elements, coordinates = read_xyz("phosphines/PdPMe3.xyz")
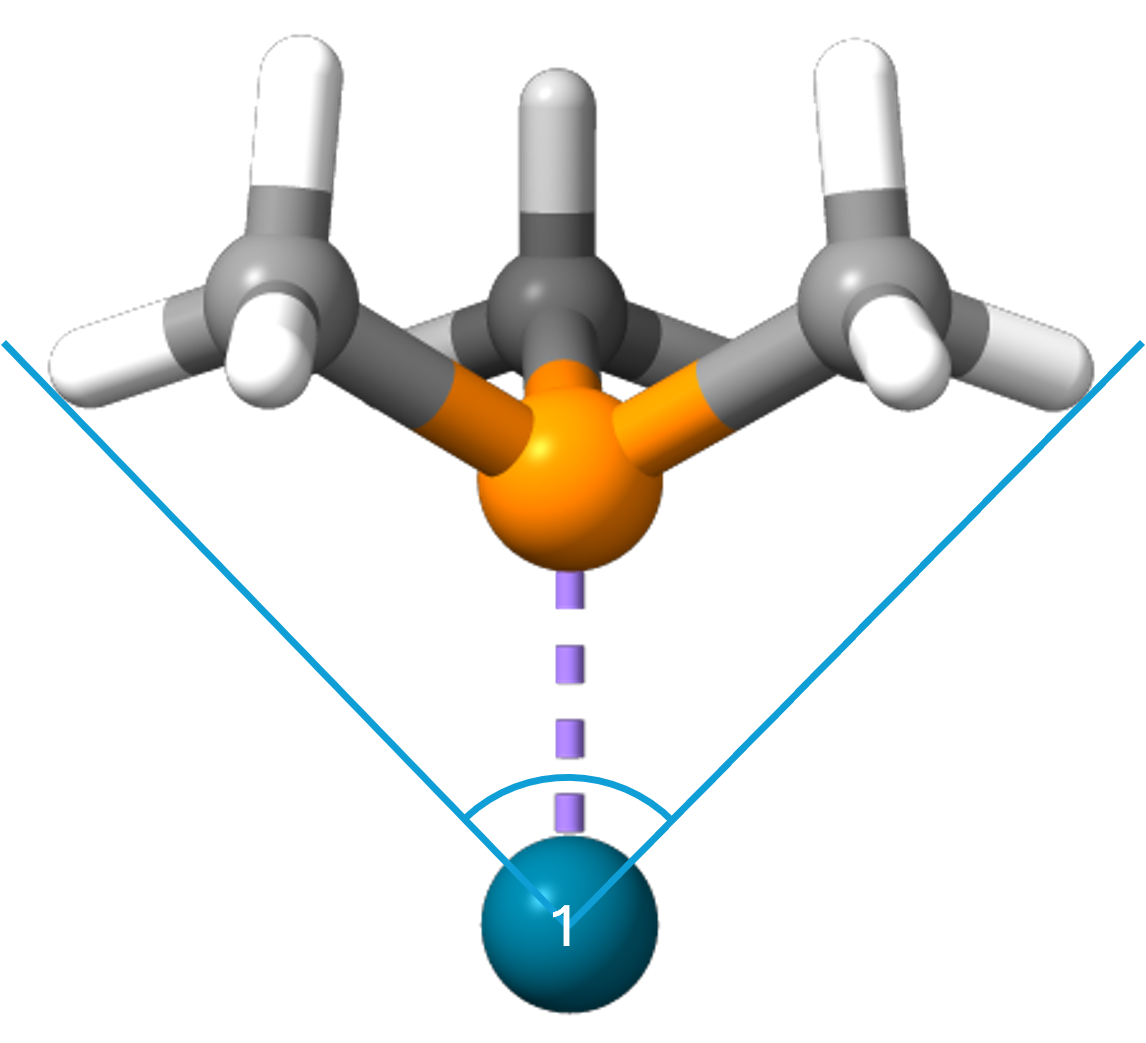
>>> cone_angle = ConeAngle(elements, coordinates, 1)
>>> print(cone_angle.cone_angle)
117.11012922937584
>>> print(cone_angle.tangent_atoms)
[5, 9, 12]
>>> cone_angle.print_report()
Cone angle: 117.1
No. tangent atoms: 3
>>> cone_angle.draw_3D()
The given atom index argument corresponds to the index of the central atom (1-indexed), Pd in the example above.
The Bondi vdW radii are used in reference [1], but
radii from the CRC Handbook is the default here. It can be changed with
radii_type=<str> with either crc or bondi. Custom radii can passed
with radii=<list>.
The default setting is method="libconeangle", which uses the fast
libconeangle package as a backend. If it is not installed, an internal
algorithm will be used (method="internal"), printing a warning.
For more detailed information, use help(ConeAngle) or see the API:
ConeAngle.
Command line script¶
The command line script provides access to the basic functionality through the terminal.
$ morfeus cone_angle PdPMe3.xyz - 1 - print_report
Cone angle: 117.1
No. tangent atoms: 3
Tangent to: H6 H10 H13
Background¶
Cone angle is a method invented by Tolman for assessing the steric size of ligands [2]. The original Tolman cone angles for phosphines have problems with asymmetric ligands and are not implemented in this package. Instead, the exact cone angles [1] are used. These are also defined for multidentate ligands.
The method implemented in ᴍᴏʀғᴇᴜs is taken directly from the article by Allen [1]. The results have been benchmarked against the original article and agree within numerical accuracy.
References